QualitySNPng builds on the principles of SNP detection as described in the QualitySNP pipeline (Tang et al, BMC bioinformatics 2006,7:438). It is optimised for the analysis of next generation sequencing data and it features:
-a user-friendly interface for SNP calling and visualisation
-multiple filtering options to handle typical sequencing errors
-support for SAM and ACE formatted input files
-exportable marker lists for the creation of SNP arrays
-visualisation of SNPs per sample (read groups)
It runs on Windows, Mac OS X and Linux, and can be used as a stand-alone application with a graphical user interface or as part of a pipeline system like Galaxy. The output data is saved as CSV text files and can be reloaded for later analysis using QualitySNPng, or processed by custom scripts for further analysis. QualitySNPng was written in C++ using the Qt toolkit.
QualitySNPng was published in the 2013 webserver issue of Nucleic Acids Research:
"QualitySNPng: a user-friendly SNP detection and visualization tool"
Harm Nijveen, Martijn van Kaauwen, Danny G. Esselink, Brechtje Hoegen and Ben Vosman
For further information please email: qsnpng @ bioinformatics.nl
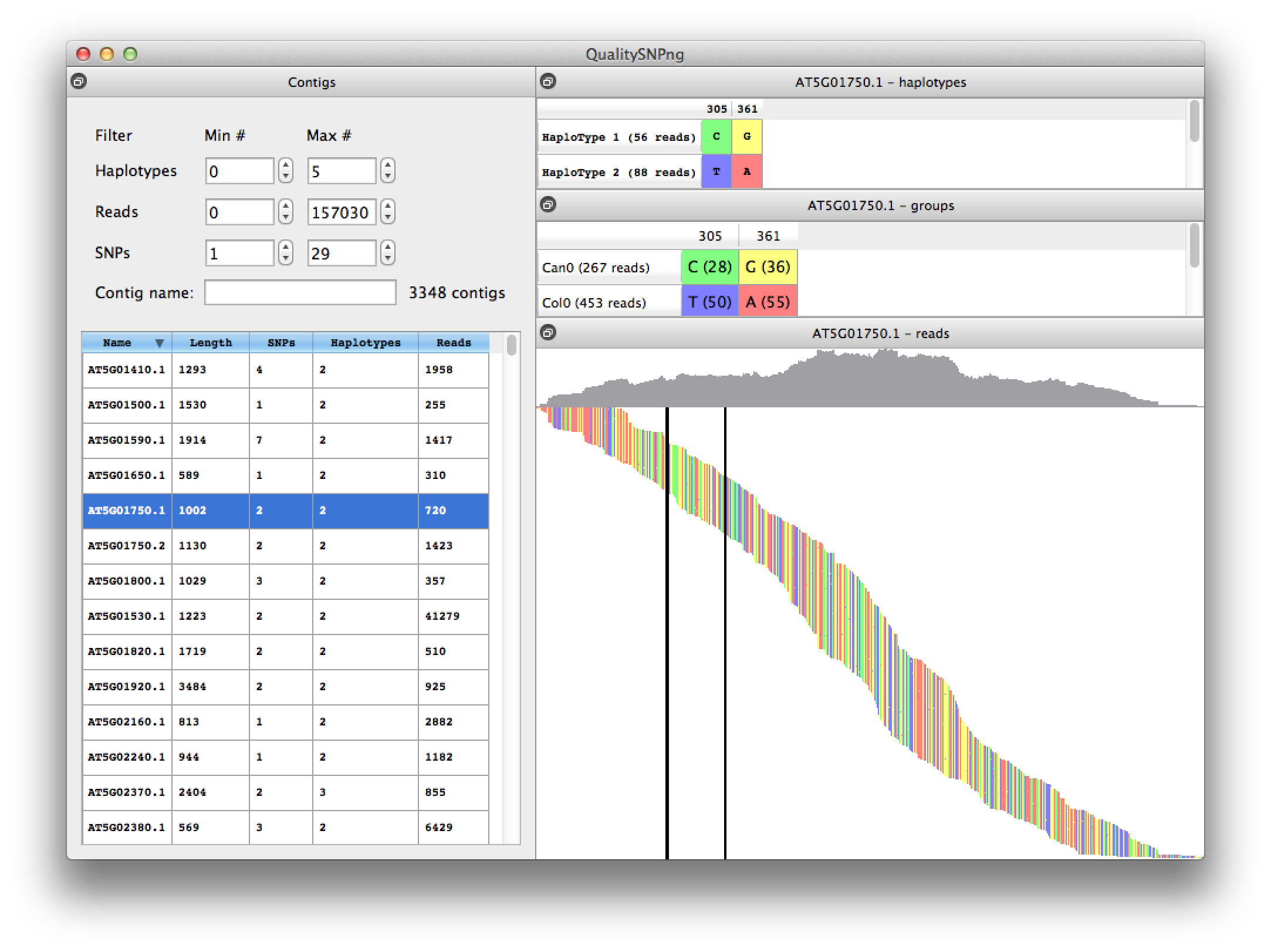
|
Figure 1 Viewing the result of a SNP analysis comparing Arabidopsis thaliana accessions Col0 and Can0 using transcriptome data from "Multiple reference genomes and transcriptomes for Arabidopsis thaliana" by Gan et al, Nature 2011.
On the left the results for the different transcripts are listed, showing the number of reads and predicted numbers of SNP and haplotypes. The list can be filtered, i.e. to show only the transcripts with a certain number of haplotypes, or using (part of) the transcript name. On the top right the predicted haplotypes are shown, which are nicely confirmed by the actual alleles per accession that are listed in the middle right table (read counts between parentheses). In the bottom right pane the reads are visualised ordered by their starting position on the transcript. The mountain landscape at the top represents the coverage per position. The two SNPs are indicated by the vertical bars. |