| Installation |
| Running QualitySNPng |
| Viewing the Results |
| Server mode |
| SNP calling algorithm |
| Run time |
| Compiling |
| Configuration file parameters |
Installation
QualitySNPng does not require additional installation steps after downloading, Mac OS X users may want to copy the program to their Applications folder. The program does expect to be able to create a directory ".QualitySNPng" in the user's home directory for new configuration files. The default output directory for results and logging is "/tmp" on Linux and OS X, and the user's temp directory on Windows systems.
Running QualitySNPng
SNP detection run
A new SNP detection run can be started by selecting "Run QualitySNPng" from the "Run" menu. This will bring up a dialog (figure 1) that allows you to provide an input (contig) file, set the output directory and a preset configuration, the configuration can be modified using the "Edit..." button. Clicking the "Run" button will start the SNP detection, the logging window will show the progress (figure 2). The "Stop" button will stop the current run and show the results that were optained thus far. When the run has finished the "Stop" button will change to "Show" to bring up the results (figure 3).
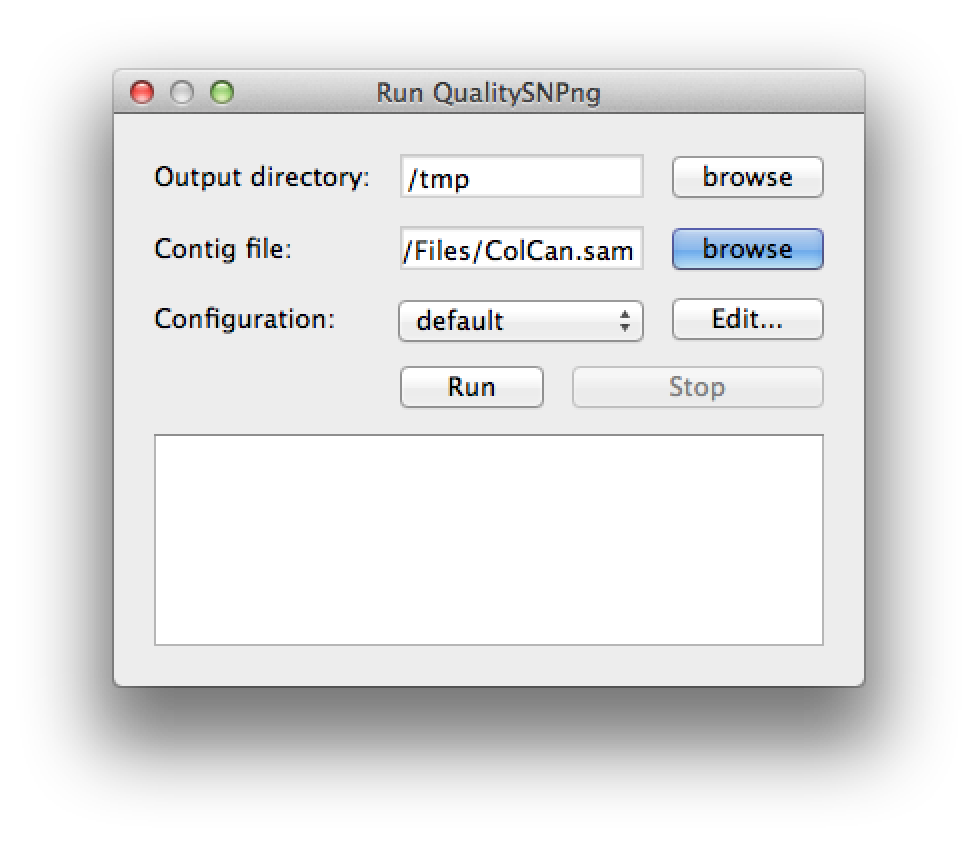 Figure 1. the run dialog |
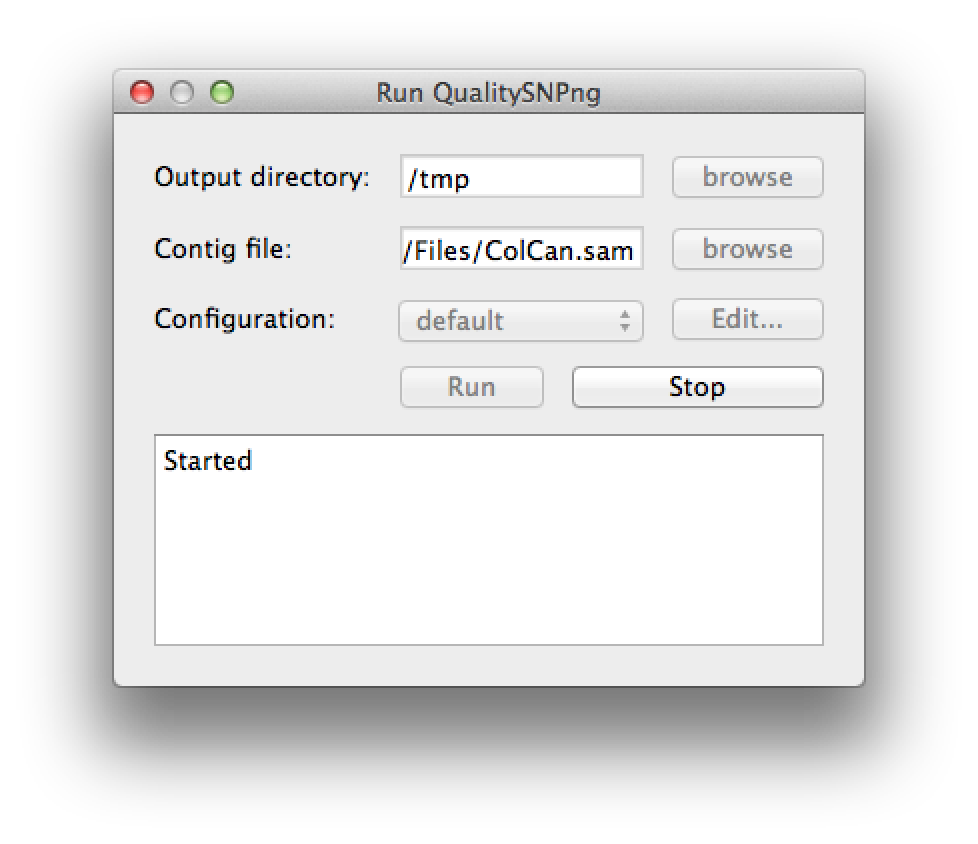 Figure 2. starting a run |
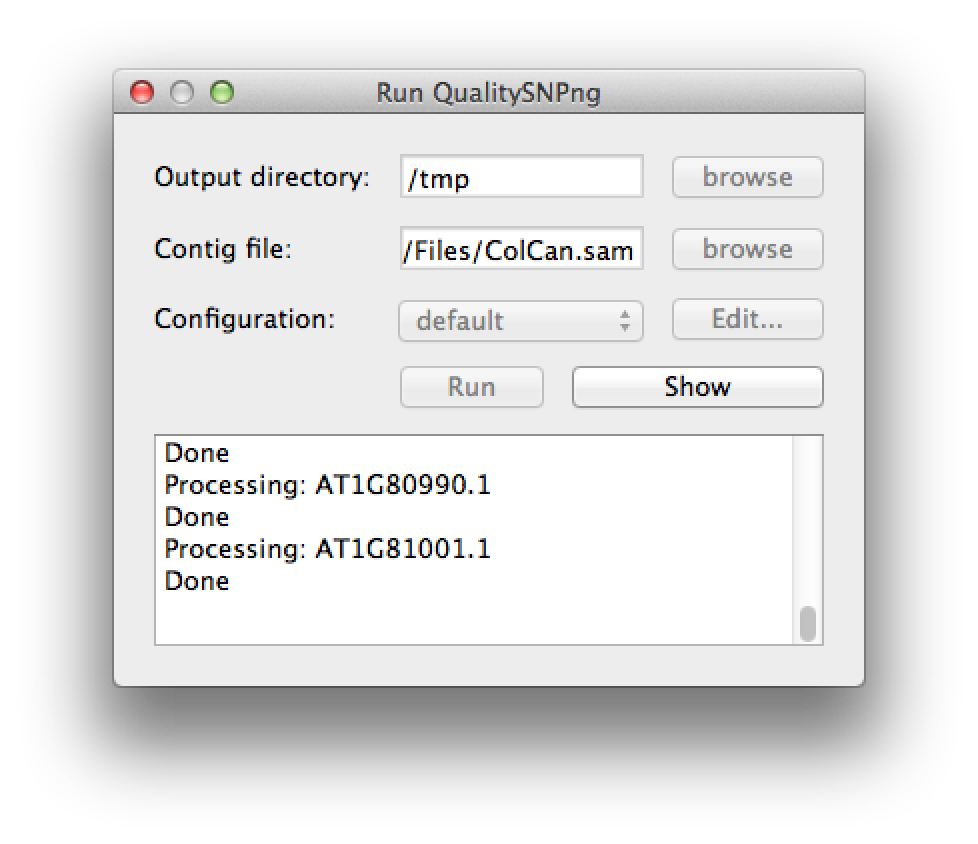 Figure 3. the run has finished |
Configuration
Pressing the edit button on the run dialog brings up the current configuration (figure 4) and allows you to change the parameters for SNP detection, haplotype prediction, marker selection and read group identification. After pressing "Ok" the changes will be saved to a configuration with the name that you provided. A configuration is saved to a file with the name of the configuration in the ".QualitySNPng" directory in the user's home directory. The settings are explained in the table following figures 4 and 5.
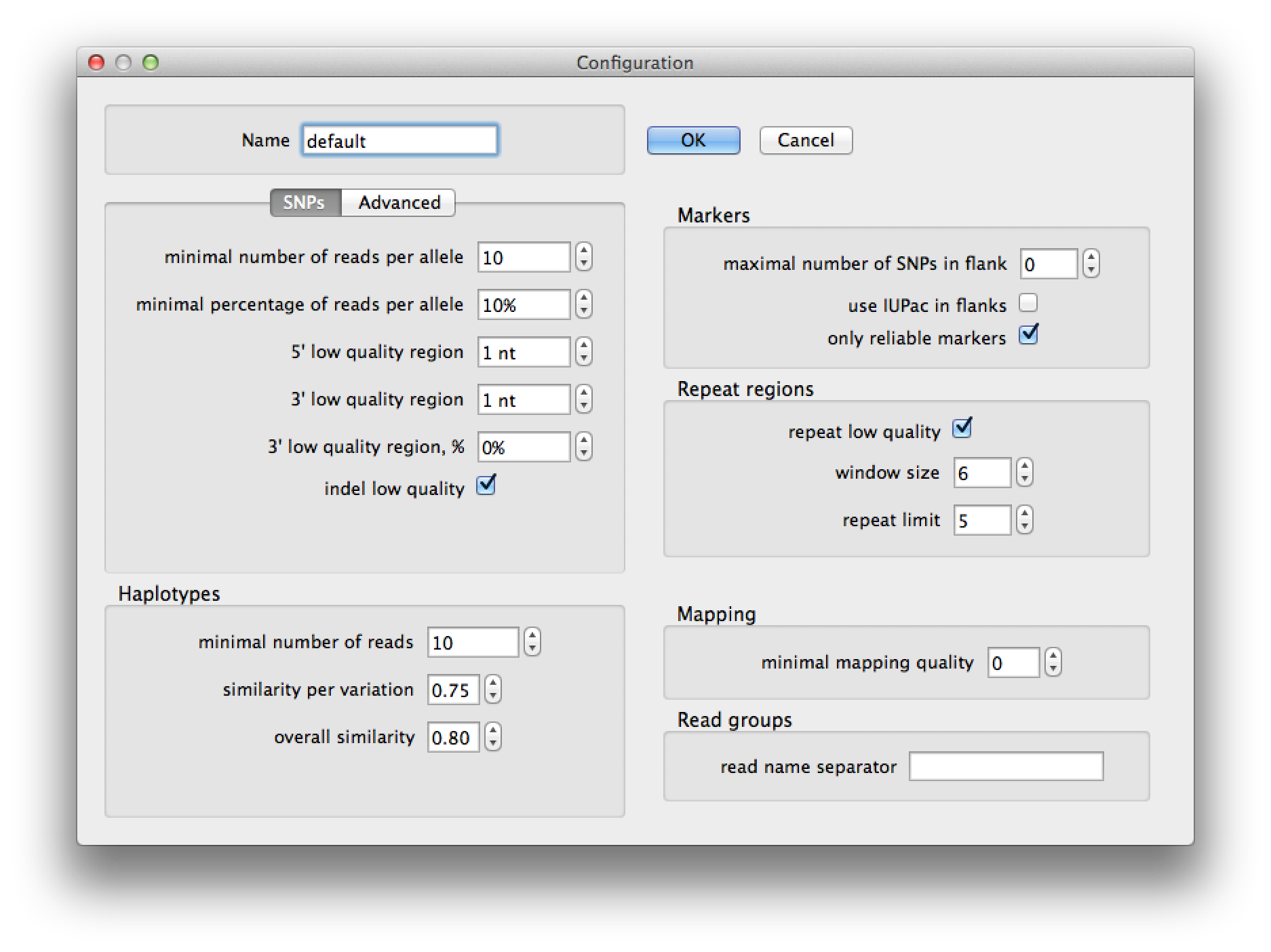 Figure 4. configuration dialog |
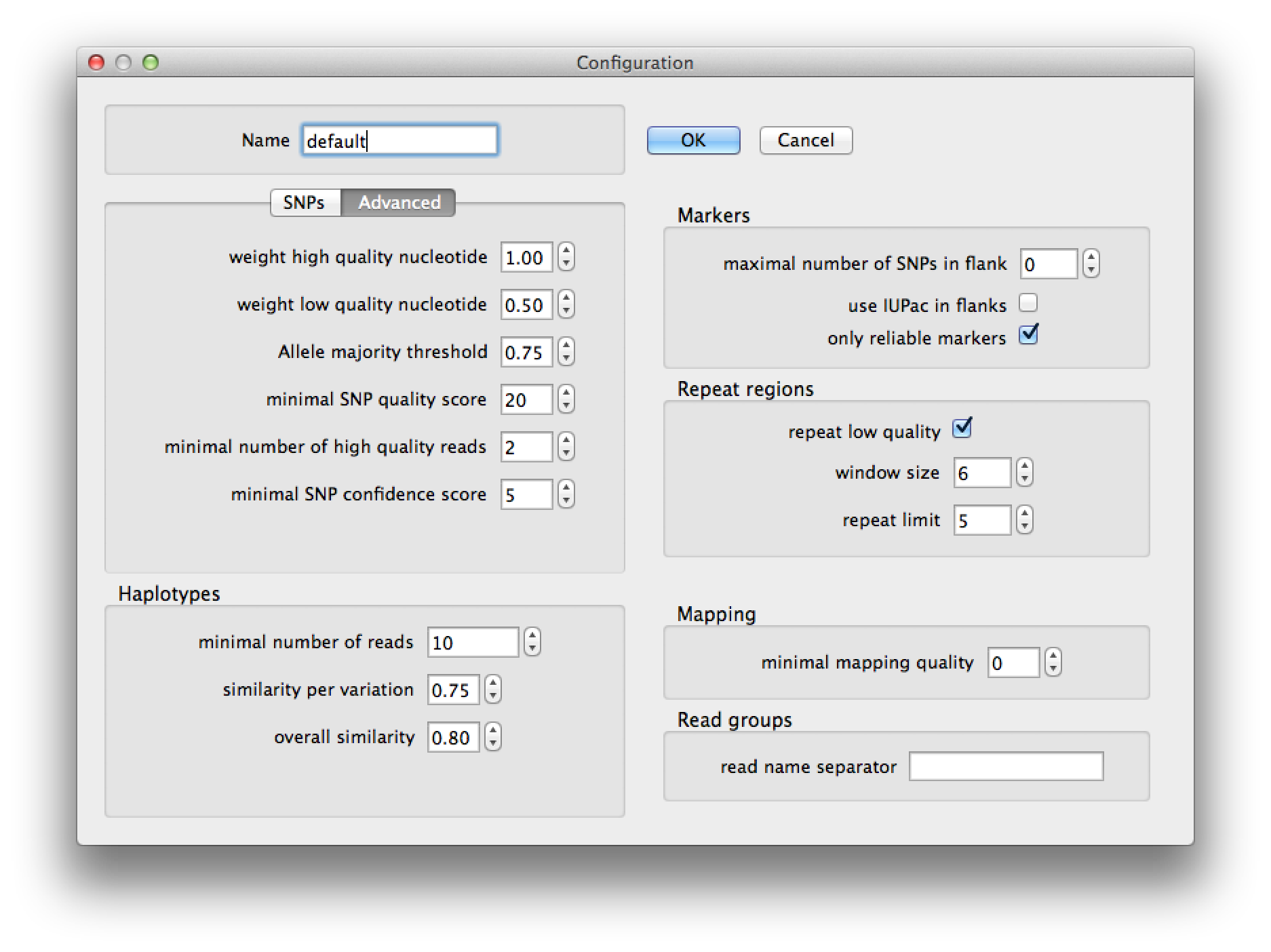 Figure 5. configuration dialog, advanced tab |
SNPs | |
| minimal number of SNPs per allele | Minimal number of reads that need to have the minor allele for a variation |
| minimal percentage of SNPs per allele | Minimal fraction of the reads that carry the minor allele |
| 5' low quality region | Number of nucleotides at the 5' end of the read that should be considered low quality. |
| 3' low quality region | Number of nucleotides at the 5' end of the read that should be considered low quality. |
| 3' low quality region, % | Number of nucleotides at the 3' end of the read as a fraction of the total read length, that should be considered low quality. |
| indel low quality | Consider indels as low quality regions. |
Haplotypes | |
| minimal number of reads | Minimal number of reads for a haplotype to be valid |
| similarity per variation | Minimal similarity score per polymorphic site, this is the threshold for the Sij variable in formula 2 in the original QualitySNP paper by Tang et al. |
| overal similarity | Minimal similarity score over all polymorphic sites, this is the threshold for the Si variable in formula 1 in the original QualitySNP paper by Tang et al. |
Markers | |
| maximal number of SNPs in flank | maximal number of SNPs allowed in flanking regions for marker SNPs |
| use IUPAC in flanks | translate SNPs in the flanking sequences of marker SNPS to their corresponding IUPAC code |
| only reliable markers | only use the reliable SNPs as marker SNPs |
Repeat regions | |
| repeat low quality | mark SNPs in low complexity regions as low quality |
| window size | the size of the window in nucleotides to check for low complexity |
| repeat limit | if one nucleotide occurs this many times in the above specified window, the region is classified as low quality |
Mapping | |
| minimal mapping quality | the minimal mapping quality a read has to have to be taken into account |
Read groups | |
| read name separator | Character(s) that were used to include the ‘read group’ name in the read name. I.e. if the separator would be __ then from a read name like: "RG1__readname" the read group would be "RG1" |
Advanced SNP | |
| weight high quality nucleotide | variable wh in formulas 4 and 5 for determining the major and minor haplotype scores in the original QualitySNP paper by Tang et al. |
| weight low quality nucleotide | variable wl in formulas 4 and 5 for determining the major and minor haplotype scores in the original QualitySNP paper by Tang et al. |
| allele majority threshold | the value 0.75 in formulas 4 and 5 for determining the major and minor haplotype scores in the original QualitySNP paper by Tang et al. |
| minimal SNP quality score | minimal Phred quality score a nucleotide has to have to be regarded as high quality |
| minimal number of high quality reads | minimal number of reads that have a high quality nucleotide at a position for that position to be regarded as high quality |
| minimal SNP confidence score | minimal confidence score for a SNP to be regarded as high confidence, see figure 2 in the original QualitySNP paper by Tang et al. |
Viewing the results
Pressing the "Show" button after a SNP detection run will bring up the results for that run (figure 6). The list can be filtered on the number of SNPs, haplotypes, reads and on (part of) the contig name. At the start the filtering is set to only show contigs with SNPs.Runs are automatically saved to the selected output directory, a new run will overwrite an existing run in the same directory. Previous runs can be loaded by choosing "Load saved run" from the "File" menu and selecting the directory the results were saved in. When QualitySNPng is started it will look for the results of a previous run in the default folder ("/tmp" on Linux and OS X, "c:\temp" on Windows systems) and load these results for viewing.
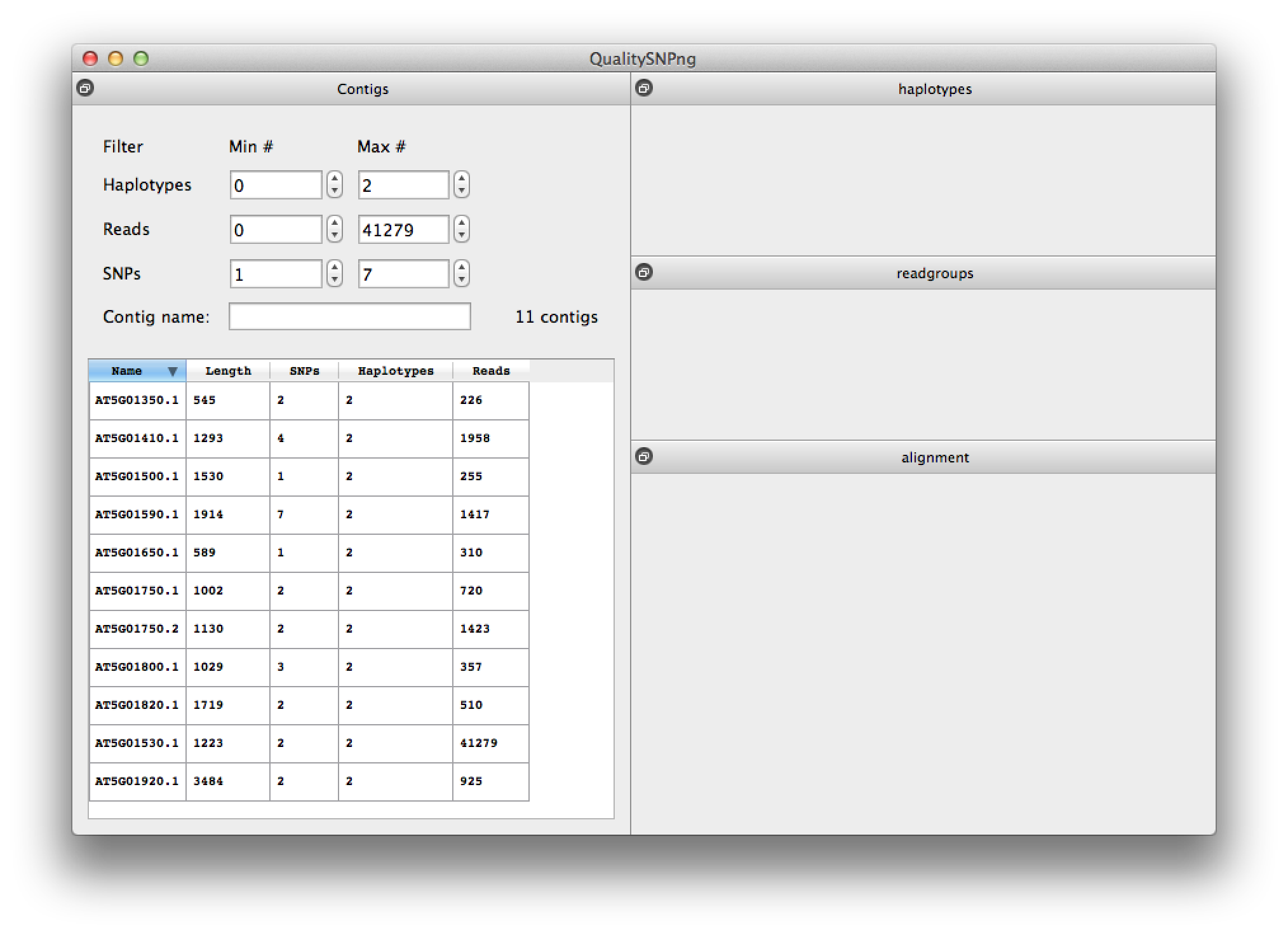 Figure 6. list of results per contig. |
Selecting a contig from the list will bring up the SNPs and predicted haplotypes for that contig (figure 7). In the top right table the predicted haplotypes are listed and in the middle right table the alleles for the different input datasets, in this case RNA-seq from the Arabidopsis thaliana accessions Col0 and Can0. Data from "Multiple reference genomes and transcriptomes for Arabidopsis thaliana" by Gan et al, Nature 2011. The predicted haplotypes nicely agree with the allele distribution for the two accesssions.
The bottom right panel shows the reads ordered by their starting position, with the coverage plotted at the top of the picture. Double clicking zooms the picture, "+" and "-" zoom in and out respectively. The "a", "s", "z" and "x" keys zoom in or out horizontally or vertically. The "f" key shows the full alignment. When zoomed in, the alignment can be panned using the mouse or arrow keys on the keyboard. Clicking a column in the haplotype or read groups tables brings the corresponding position in view. The "h" key can be used to reset the left top corner of the alignment. A mouse-over shows information about the read that is currently under the mouse pointer.
The different windows can be undocked, reordered and resized individualy to make optimal use of the available screen real estate.
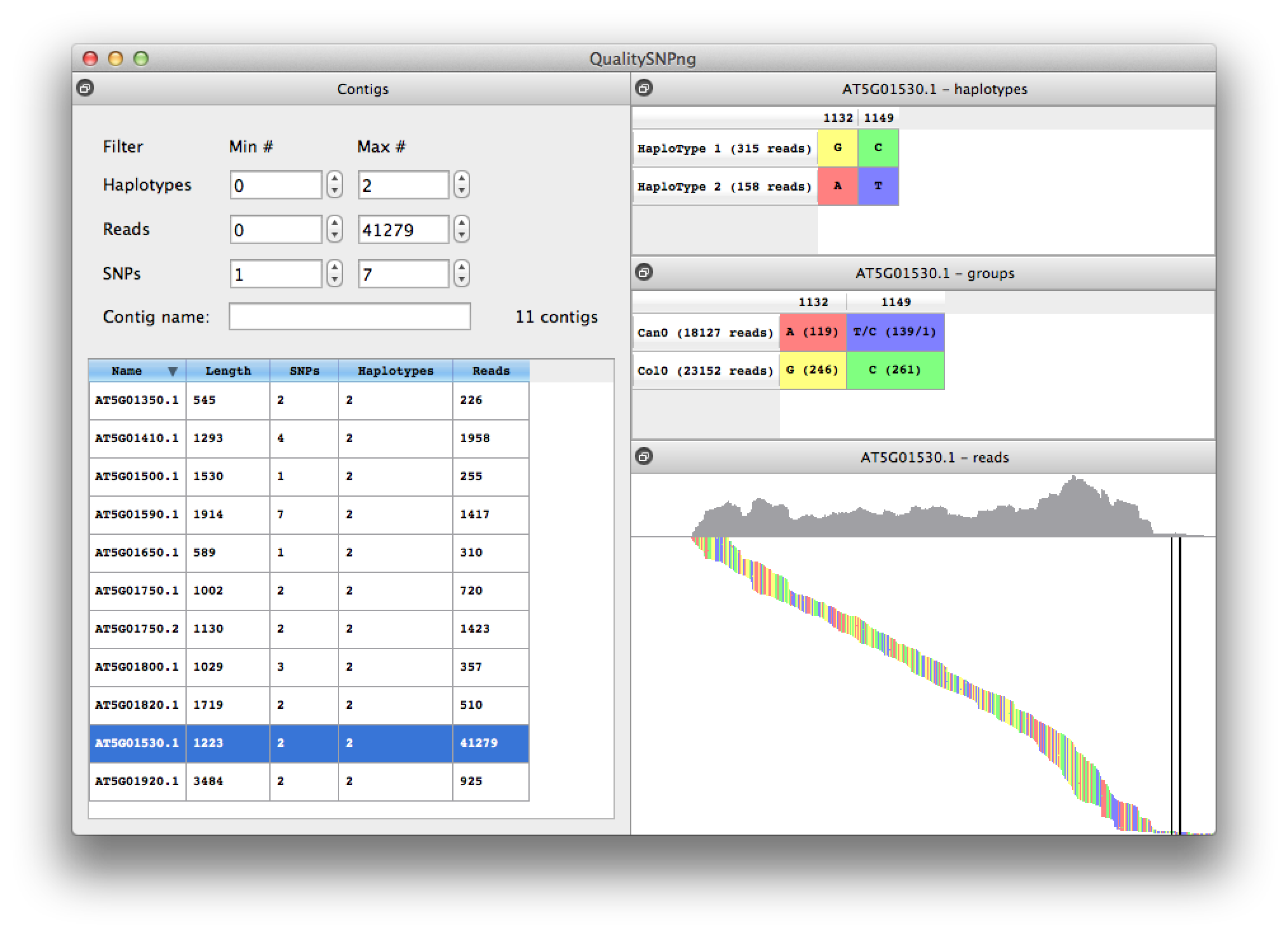 Figure 7. details for one contig |
When two adjacent SNPs are further apart than the length of a read, the haplotype prediction algorithm cannot link the SNPs into one haploype and will show a new haplotype (figure 8). Because for this experiment we have accession information availble, from the information in the middle right table it is clear that haplotypes 1 and 3 belong together, as well as haplotypes 2 and 4.
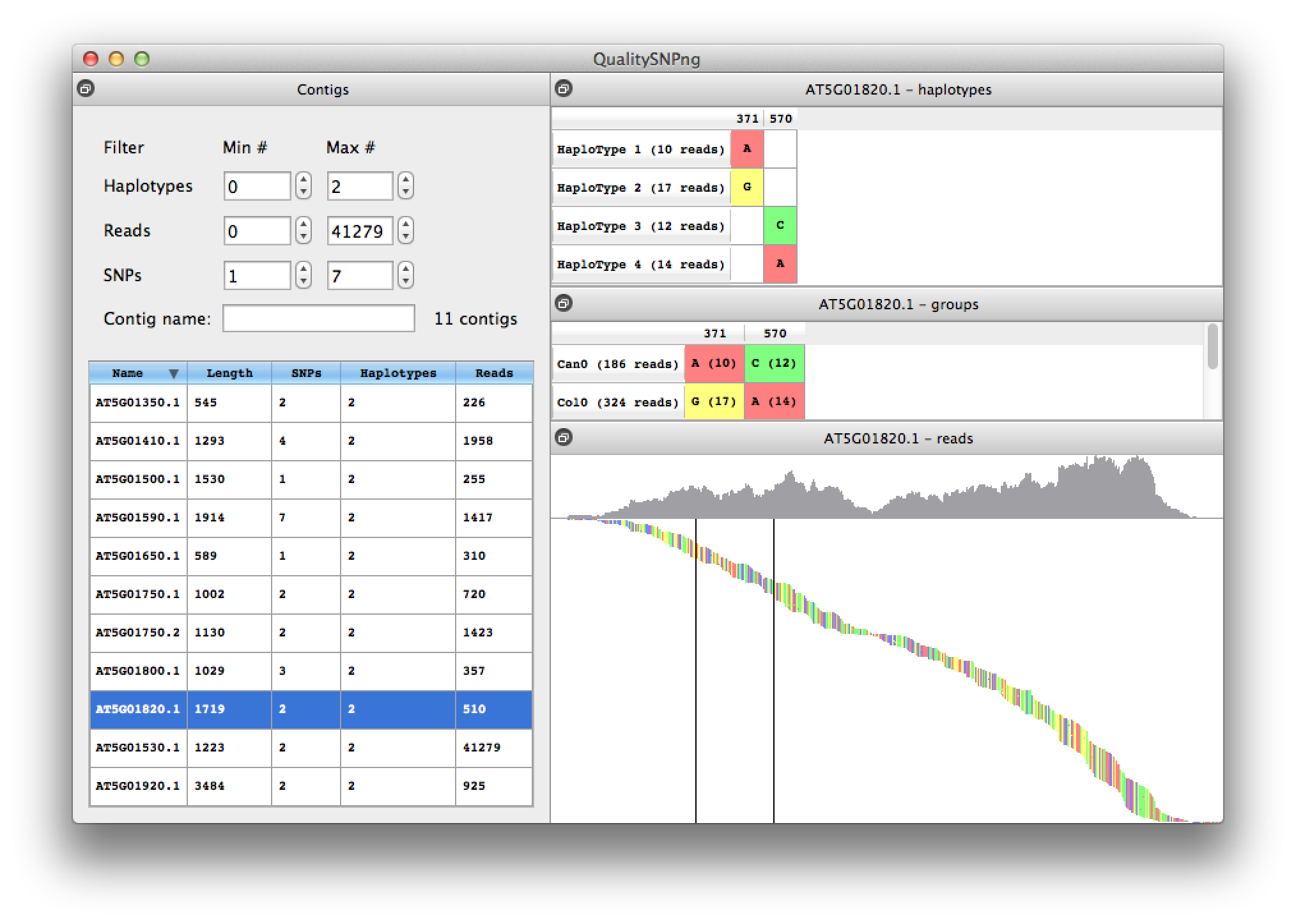 Figure 8. contig with two split haplotypes |
With the right mouse button the sorting of the reads can be changed from on starting position to haplotype, to show the reads grouped per haplotype (figure 9). Using the read information available in the mouse-over tooltips confirms that indeed the reads in haplotypes 1 and 3 are from Can0 and the reads in haplotypes 2 and 4 from the Col0 accession. The reads at the bottom of the alignment were not assigned to a haplotype since they do not contain informative SNPs.
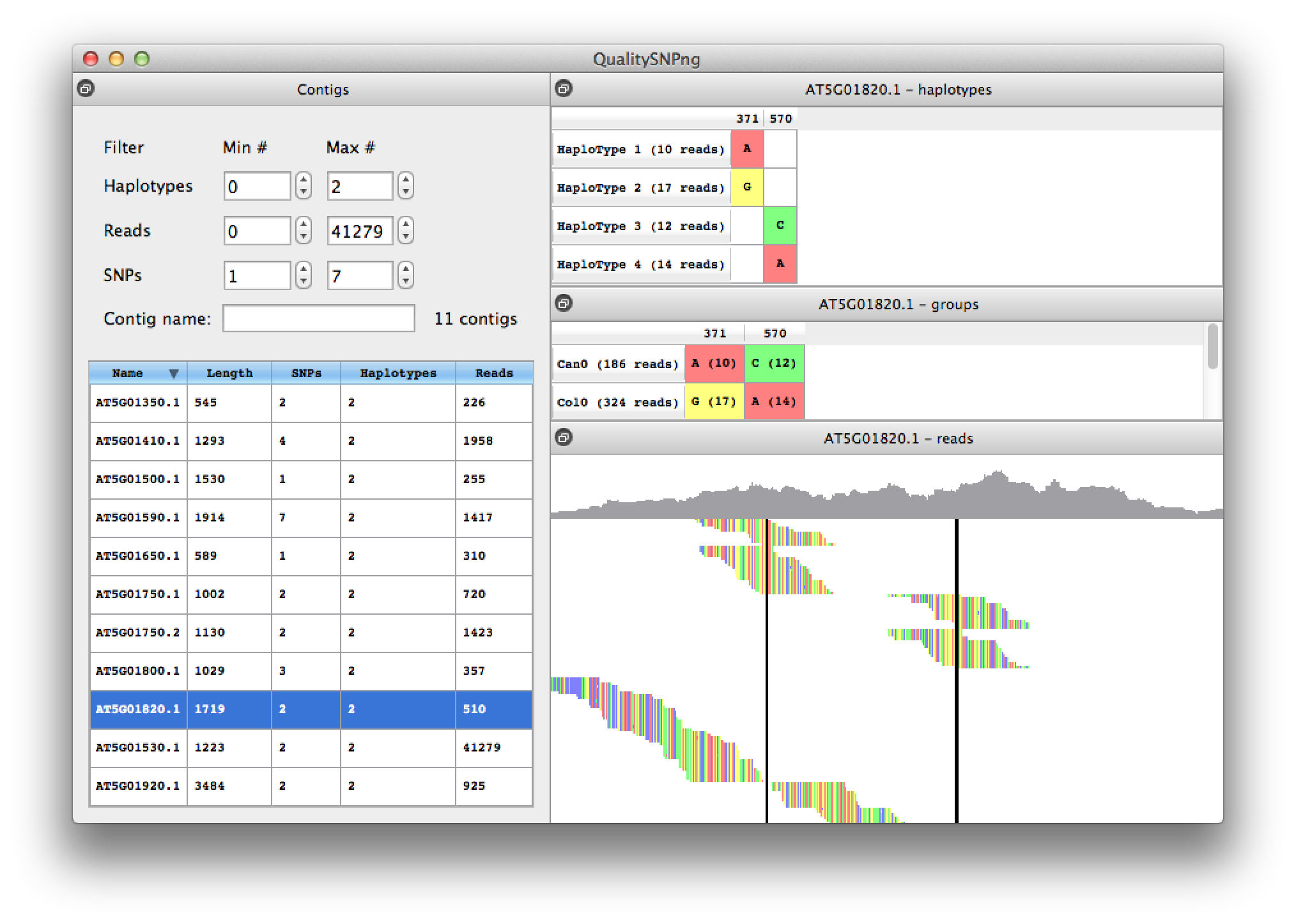 Figure 9. reads sorted on haplotype |
By selecting the "Export marker list" from the "File" menu, you get a list of all available SNPs that can be used as markers, for instance in genotyping assays. Setting the flank size filters for SNPs that have a flanking sequence both upstream and downstream of high quality nucleotides. The list is affected by the filtering options for the contig list. The marker SNP it self is marked with square brackets listing the major and minor allele. The list can be exported to a CSV file for further processing, and can easily be imported in a spreadsheet program like Excel.
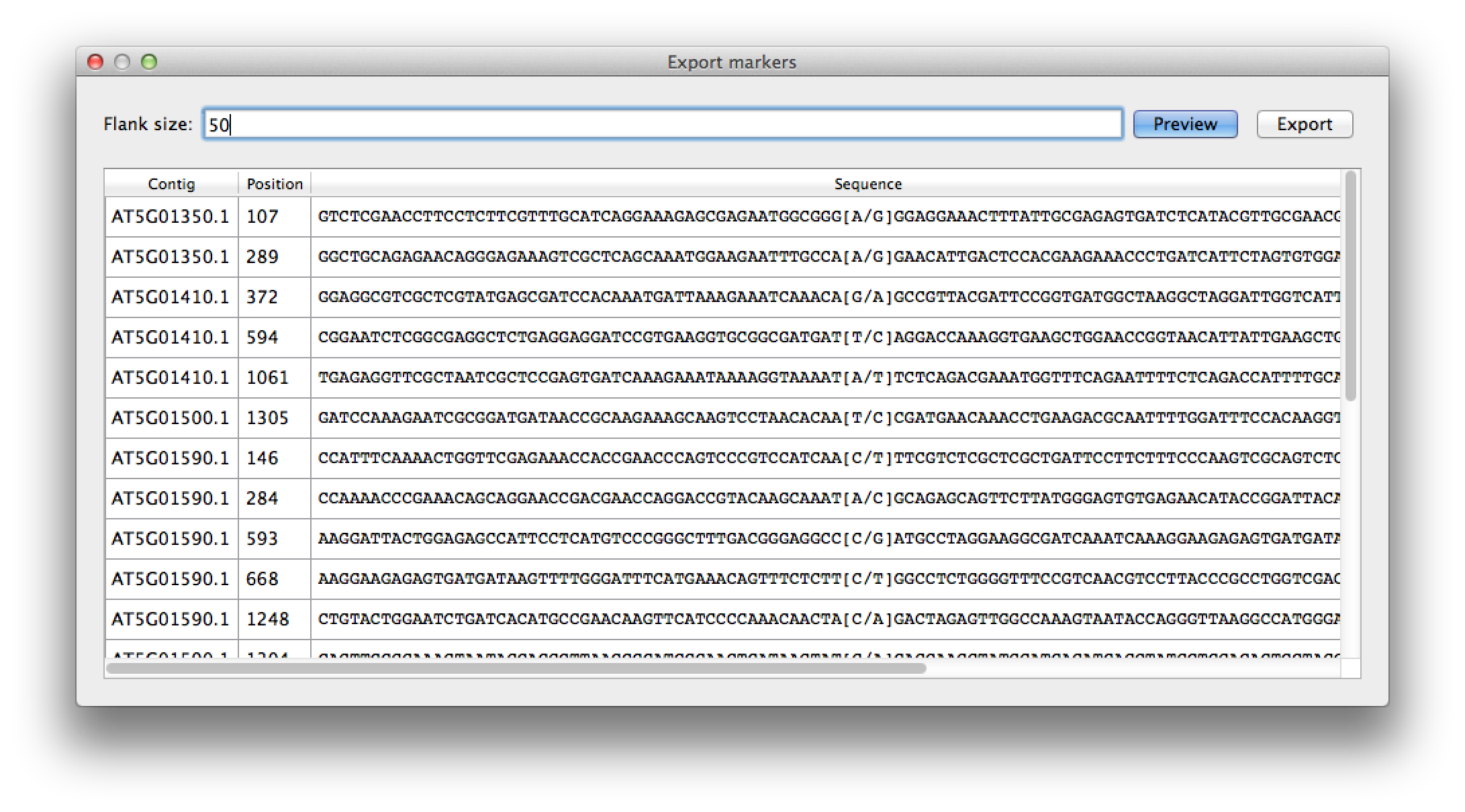 Figure 10. marker selection, with flank size of 50 nt. |
Server mode
Using the "-servermode" argument turns QualitySNPng into a commandline tool without graphical interface. This allows QualitySNPng to be used as part of an analysis pipeline, like in Galaxy, or stand alone remote on a compute server when the expected run time is several hours. The settings for the SNP calling run can be provided in a configuration file.
Example:
./QSNPng -servermode -config athaliana.cfg ColCan.sam
The QualitySNPng configuration files have one parameter setting per line with this format: <parameter>=<value>
Example:
minimalNumberOfReadsPerAllele=10
For all available parameters, see the Settings section below. The configuration files that are created when making changes to the configuration dialog are saved in the directory the QualitySNPng program is located it. These files can be used in server mode as well.
QualitySNPng produces text output files for the reads, contigs and the predicted variations and haplotypes. One row in these files represents a read, contig, variation or haplotype, with the data fields in different columns. The first row of each file lists the column headers. These output files can be used for further processing, for instance when QualitySNPng is used as part of an analysis pipeline. Alternatively the result files can be viewed with QualitySNPng by opening the directory they are in.
SNP calling algorithm
QualitySNPng takes as input a sequence alignment in the form of a (sorted) SAM (Li et al. 2009) or ACE (Gordon et al. 1998) formatted file with single-end or paired-end reads and then employs three filtering steps to detect reliable variations.The first step labels all nucleotide differences that occur in at least a configurable number or fraction of the reads as potential SNPs.
The second filtering step takes into account the quality of the sequence region containing the variant nucleotide and selects only the high confidence SNPs. The sequence quality depends on the Phred score (Ewing et al., 1998) if available, and additionally can be modified based on specific sequence patterns. Variations found in homopolymeric tracts can be set to be of low quality, if these are known to be prone to errors by the used sequencing technology, or alignment program. Also a number of nucleotides at the 5' or 3' ends can be labelled as low quality, for instance to avoid false SNPs caused by incomplete adaptor trimming.
In the third filter step haplotypes are predicted based on the high confidence SNPs. Only if a variation is supported by one or more haplotypes it is considered to be a reliable SNP. These three filters are similar to those that are used by the original QualitySNP software (Tang et al. 2006), except that the second and third filters were reversed to make sure that the detected haplotypes are based on high confidence SNPs.
Run time
The run time largely depends on the size and nature of the input sequencing data, ranging from less than a minute for a set of ~25,000 contigs (~100 reads/contig), to 10 minutes for one large single contig of 7,000 bp with 800,000 reads. Larger and more variable sequence alignments can take longer, also depending on the stringency of the settings: lowering the threshold for potential SNPs will result in more work for the second and third filters that are computationally the most expensive.For large input files that are expected to take several hours to process one could use the tool in "servermode" to perform the SNP calling on a compute server and subsequently analyse the results using the GUI.
Configuration file parameters
minimalNumberOfReadsPerAllele (integer value, default 2) Minimal number of reads that need to have the minor allele for a variationminimalNumberOfReadsPerAllelep (real value between 0 and 1, default 0.1) Minimal fraction of the reads that carry the minor allele
minimalNumberOfReadsPerHaploType (integer value, default 2) Minimal number of reads for a haplotype to be valid
lowQualityRegion5prime (integer value, default 0) Number of nucleotides at the 5' end of the read that should be considered low quality. In the first version of QualitySNP for Sanger reads this was set to 30.
lowQualityRegion3prime (integer value, default 0) Number of nucleotides at the 3' end of the read that should be considered low quality.
lowQualityRegion3primePerc (real value between 0 and 1, default 0.0) Number of nucleotides at the 3' end of the read as a fraction of the total read length, that should be considered low quality. In the first version of QualitySNP for Sanger reads this was set to 0.2 (20%).
maxNumberOfSNPsInFlanks (integer value, default 0) Allowed number of SNP in the flank of a potential marker SNP.
weightHighQualityRegion (real value between 0 and 1, default 1.0) variable wh in formulas 4 and 5 for determining the major and minor haplotype scores in the original QualitySNP paper by Tang et al.
weightLowQualityRegion (real value between 0 and 1, default 0.5) variable wl in formulas 4 and 5 for determining the major and minor haplotype scores in the original QualitySN P paper by Tang et al.
minSNPQualityScore (integer value, default 20) Phred quality score cut-off for the individual read quality for a certain nucleotide.
minimalConfidenceScore (integer value, default 2) Confidence score cut-off for a SNP to be considered high confidence.
minNumberOfHighQualityReads (integer value, default 2) Minimal number of high quality reads required to produce a high confidence SNP
logLevel (default QSNP_WARNING) Logging level can be set to info, warning, error, debug
minimalMappingQuality (integer value, default 0) Reads with a mapping quality below this value are discarded.
maxNumberOfReads (integer value, default 0) QualitySNPng will consider at most this number of reads per contig and disregard the rest. Can be used to speed up analyse for datasets with a surplus of reads. Setting the value to 0 effectively disables this setting.
lowComplexityRegionSize (integer value, default 5) Window size for detecting low complexity regions
lowComplexityRepeatCount (integer value, default 4) Number of identical nucleotides within a window that flags a region as low complexity, making SNPs that fall in that area low confidence.
similarityPerPolymorphicSite (real value between 0 and 1, default 0.75) Minimal similarity score per polymorphic site, this is the threshold for the Sij variable in formula 2 in the original QualitySNP paper by Tang et al.
similarityAllPolymorphicSites (real value between 0 and 1, default 0.8) Minimal similarity score over all polymorphic sites, this is the threshold for the Si variable in formula 1 in the original QualitySNP paper by Tang et al.
alleleMajorityThreshold (real value between 0 and 1, default 0.75) the value 0.75 in formulas 4 and 5 for determining the major and minor haplotype scores in the original QualitySNP paper by Tang et al.
outputDirectory (default "/tmp") Path of the output directory.
inputDirectory (default "/tmp") Path of the input directory, i.e. the directory where the configuration file and the contig file would be searched for.
contigFileName (default empty) Name of the file with the contigs (SAM or ACE format).
logFileName (default "QSNP.log") Name of the log file.
contigsFile (default "contigs.csv") Name of the output file for contigs.
readsFile (default "reads.csv") Name of the output file for the reads.
haploTypesFile (default "haplotypes.csv") Name of the output file for the haplotypes.
variationsFile (default "variations.csv") Name of the output file for the variations.
readGroupsFile (default "readgroups.csv") Name of the output file for the read groups.
readNameGroupSeparator (default empty) Character(s) that were used to include the ‘read group’ name in the read name. I.e. if the separator would be __ then from a read name like: "RG1__readname" the read group would be "RG1".
configurationFile (default empty) Filename and path for the configuration file
fieldSeparator (default tab) Column separator to be used for the output files
indelLowQuality ((true (1) or false (0), default true) Consider indels as low confidence
lowComplexityLowQuality ((true (1) or false (0), default true) Consider SNPs in a low complexity region as low confidence
printAlignment ((true (1) or false (0), default false) Print a crude alignment of the reads to the terminal, useful for testing purposes
printHaploTypes ((true (1) or false (0), default false) Print an overview of the reported haplotypes to the terminal, usefull for testing purposes
printSNPs ((true (1) or false (0), default false) Mark the SNPs on the alignment, only works together with printAlignment
printMarkers ((true (1) or false (0), default false) Show the markers on the alignment, only works together with printAlignment
printSummaryLine ((true (1) or false (0), default true) Print out a summary line for each contig to the terminal, usefull for testing purposes
useIUPACCodes ((true (1) or false (0), default false) For marker SNPs, instead of the reference nucleotide print the IUPAC code for SNPs that occur in the flanking sequences
onlyReliableMarkers ((true (1) or false (0), default true) Only use reliable SNPs as marker SNP
showContigsWithoutSNP ((true (1) or false (0), default true) If set to false, contigs without SNPs will not be included in the output
collectStatistics ((true (1) or false (0), default true) If set to false, QualitySNPng will not go throught the input file at the start to collect statistics about the number of contigs and read groups
outputReadGroups ((true (1) or false (0), default true) If set to false, no read group information will be reported
servermode (true (1) or false (0), default false(0) ) Flag to turn on the command line mode without a graphical interface.
Compiling
QualitySNPng can be compiled from source using Qt Creator that is available for Windows, Mac OS X and Linux. The source was developed using Qt version 4.7.4. After downloading the source, open the QualitySNPng.pro file with QtCreator.