HaploSNPer Manual
Index
1. Introduction of
HaploSNPer
2. How to use the program
3. How to use the
results
1. Introduction of HaploSNPer
HaploSNPer is a flexible web-based tool for detecting
haplotypes and single nucleotide polymorphisms (SNPs) in user-specified input
sequences from both diploid and polyploid species. It
includes BLAST for finding homologous sequences in the species-specific EST
databases, CAP3 or PHRAP for aligning them, and QualitySNP for discovering
reliable haplotypes and SNPs. All possible and reliable haplotypes (which in
this context are the available alleles of a gene in the database) are detected
by a mathematical algorithm using potential polymorphism information. Reliable
SNPs are then identified based on the reconstructed haplotypes and sequence
redundancy.
2. How to use the program
HaploSNPer is a friendly and flexible tool, it can be
run in two possible ways, either interactive process: users submit sequences
and wait for the process, or in batch mode: users submit sequences and a email
at the same time, and the links of their results will be sent to them by email
upon job completion and also a download link is supplied to save their results.
Moreover, the input option is flexible: users can
submit a sequence in FASTA format as the seed sequence, and sequences
homologous to the seed will be retrieved from the selected database; at the
same time, users can submit a number of similar or homologous sequences to the
seed sequence as well. This is especially useful when users want some
homologous sequences that are not yet available in public database to be part
for the detection of haplotypes and SNPs, and these sequences and the seed
sequence will be merged with the homologous sequences from the selected
database, and haplotypes and SNPs will be detected on the merged dataset.
Alternatively, HaploSNPer can process a dataset containing a number of
homologous or similar sequences submitted by users without specifying a database, the detection will be executed only on this
dataset.
The seed sequence is either input in the seed sequence
text area or uploaded from a file (the left part of the input option). Other
similar sequences are either input in the text area of other similar sequences
or uploaded from a file (the right part of the input option). Importantly,
these sequences should be formatted to FASTA format (see below). Parameters can
be set to tailor the performance and output of HaploSNPer to the specific
requirements of the user.
1) Format
of input sequences
The seed sequence and other similar sequences need
formatting in FASTA. Besides nucleotide information, as well as other related
information like cultivar or strain, tissue and unigene ID also can be
included. The name description of these sequences is as following:
>sequence name|genotype|tissue|unigene
ID
For example:
a. An example of the seed
sequence from apple
>AY789239|Prima|leaf|unknown
ATGGGTGTCTACACATTTGAGAACGAGTACACCTCTGAGATTCCACCACCAAGATTGTTC
AAGGCCTTTGTCCTCGATGCTGATAACCTCATCCCCAAGATTGCACCCCAGGCAATCAAG
CATGCTGAGATCCTTGAAGGAGACGGTGGCCCTGGAACCATCAAGAAGATCACTTTTGGT
GAAGGCAGCCAATACGGCTACGTGAAGCACAAGATCGACTCAGTTGACGAAGCAAACTAC
TCATACGCCTACACTTTGATTGAAGGAGATGCTTTGACAGACACCATTGAGAAGGTCTCT
TACGAGACCAAGTTGGTGGCATCTGGAAGTGGTTCCATCATCAAGAGTATCAGCCACTAC
CACACCAAGGGTGATGTTGAGATCAAGGAAGAGCACGTCAAGGCTGGCAAAGAGAAGGCT
CATGGTTTGTTCAAGCTTATTGAGAGCTACCTCAAGGGCCACCCCGACGCATACAACTAA
b. an
example of other sequences similar to the seed sequence
>AY789238|Fiesta|leaf|unknown
ATGGGTGTCTGCACATTTGAGAACGAGTTCACCTCTGAGATTCCACCATCAAGATTGTTC
AAGGCCTTTGTCCTTGATGCTGACAACCTCATCCCCAAGATTGCACCCCAGGCAATCAAG
CAAGCTGAAATCCTTGAAGGAAACGGTGGCCCCGGAACCATCAAGAAGATCACTTTTGGT
GAAGGCAGCCAGTACGGCTACGTGAAGCACAGGATTGACTCAATTGACGAAGCAAGCTAC
TCATACTCCTACACTTTGATTGAAGGAGATGCTTTGACAGACACCATCGAGAAAATATCT
TACGAGACCAAGTTGGTGGCATGTGGAAGTGGTTCCACCATCAAGAGCATCAGTCATTAC
CACACCAAGGGAAACATTGAAATCAAGGAAGAGCACGTCAAGGCTGGAAAAGAGAAGGCC
CATGGTTTGTTCAAACTTATTGAGAGCTACCTTAAGGACCACCCCGACGCATACAACTAA
>AY789240|Prima|leaf|unknown
ATGGGTGTCTACACATTTGAGAACGAGTACACCTCTGAGATTCCACCACCAAGATTGTTC
AAGGCCTTTGTCCTCGATGCTGATAACCTCATCCCCAAGATTGCACCCCAGGCAATCAAG
CATGCTGAGATCCTTGAAGGAGACGGTGGCCCTGGAACCATCAAAAAGATCACTTTTGGT
GAAGGTAGCCAATACGGCTACGTGAAGCACAAGATCGACTCGGTTGACGAGGCAAACTAC
TCATACGCCTACACTTTGATTGAAGGAGATGCTTTGACAGACACCATTGAGAAGGTCTCT
TACGAGACCAAGTTGGTGGCATCTGGAAGTGGTTCCATCATCAAGAGTATCAGCCACTAC
CACACCAAGGGTGATGTTGAGATCAAGGAAGAGCACGTCAAGGCTGGCAAAGAGAAGGCT
CATGGTTTGTTCAAGCTTATTGAGAGCTACCTTAAGGGCCACCCCGACGCATACAACTAA
Note:
1. If you don't know genotype, tissue and unigene ID of your sequences, and it is possible to use only sequence name or
sequence accession. For instance: >seq112233
2. If descriptions of the sequence name, genotype and tissue
contain spaces,then
substitute it by "-". Such as: >AB1001|apple-tree|young-leaf
3. The seed sequence and other similar sequences can also
be uploaded from simple text files after these sequences are formatted in
proper format.
4. The annotation information (cultivar/strain, genotype,
tissue and unigene) is retrieved by the accession of a sequence from EMBL
database. It means that only sequences existing in the public database EMBL or
NCBI may have the annotation information.
5. The number of similar or homologous sequences submitted
by the option of "Other similar sequences" is limited to 500. If you
want to process more than 500 sequences, please contact us: mail webmaster@bioinformatics.nl.
The input interface of HaploSNPer is as following:
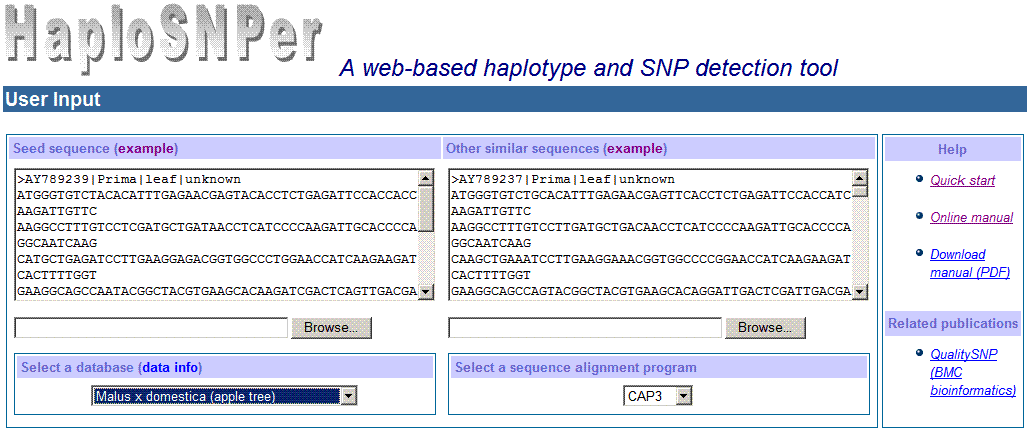
2) Settings of HaploSNPer's
Parameters
There are seven component parameters required to
control the performance of HaploSNPer, a) selecting a tagging database, b)
selecting a sequence alignment program, c) pre-processing of sequences,
d)settings for BLAST and CAP3 or PHRAP, e) settings for haplotype
reconstruction, f) settings for low quality region of sequences, g) settings
for SNP detection, h) and settings for visualizing output. Of which e, f and g
are used to control the performance of QualitySNP.
a. Selecting a tagging database
The tagging database can be selected from the list of
species-specific EST databases. These databases contain all EST sequences of
these species extracted from the EMBL database, the number of sequences in each
database and EMBL version can be checked by the link "data info".
Moreover, these databases store not only nucleotide information, but also
including cultivar or strain, tissue and unigene ID if they are available in
EMBL database. The information will be used by HaploSNPer for showing haplotypes
and SNPs.
b. Selecting a sequence alignment
program
CAP3 or PHRAP can be chosen for sequence alignment. For
SNP mining, CAP3 is recommended as it uses individual sequence overlap for
cluster constructing, while PHRAP tends to extend the consensus sequence by
overlap. However, PHRAP is much faster than CAP3.

c. Pre-processing of sequences
Many sequences
obtained from the public database were poor quality containing vector fragments.
It can make wrong sequence assembling and SNP detection. Cross_match is a good
tool to remove vector sequences according to the vector file that was
downloaded from NCBI. Besides containing vector sequences, some sequences
contain long repeat fragments, which can make wrong sequence assembling as
well. RepeatMasker is good at masking repeat fragments. The Repeat database was
downloaded from http://www.girinst.org/. Taking account into these cases,
HaploSNPer supplies Cross_match and RepeatMasker to clean sequences for users. Usually, Cross_match and RepeatMasker take long time to process
sequences. So when one of them is selected, an email address is required and
the output is returned the email address instead of waiting for a web page.
Vector file and Repeat database will be updated as their
new versions are released. 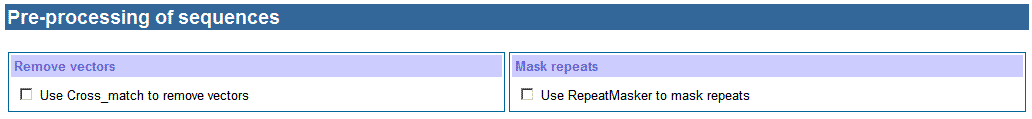
d. Settings of parameters
for BLAST and CAP3 or PHRAP
BLAST is used to search for sequences
homologous/similar to the input seed sequence. E-value for BLAST can be set to
select sequences similar to the seed sequence. When E-value is high, many
similar sequences will be found and that will produce many clusters; while E-value
is low, there will be not enough similar sequences for haplotype and SNP
detection. A series of E-values have been tested on several sequences; an
E-value of 1e-60 usually results in the selection of sequences sufficiently
similar to the seed sequence, and this value is the default in HaploSNPer.
Users can adjust it according to the results using the default settings.
For exampe, if no homologous sequences are found, users can try to increase the E-value
that may find some similar sequences; if too many clusters are produced, user can
try to decrease the setting that results in less sequence seleted.
The similarity for sequence assembling of CAP3 or PHRAP
can be set by users. According to our experience, the similarity for CAP3 and
PHRAP is 95%, which is stringent enough to prevent most paralogous sequences
and keep all available allelic sequences in a cluster.
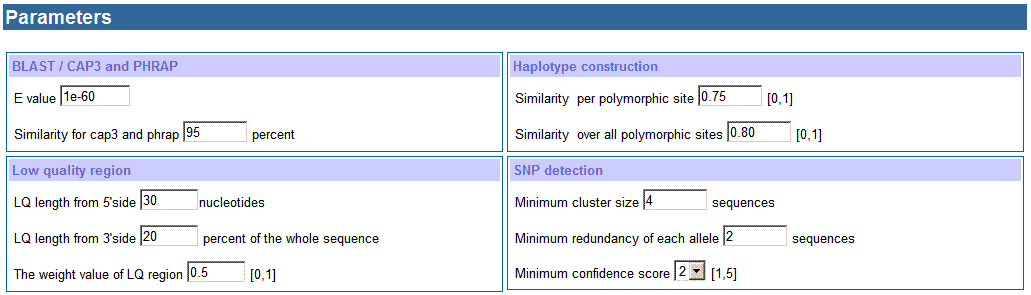
e. Settings for haplotype
reconstruction
In the QualitySNP algorithm, a potential haplotype is
defined as a group of sequences within a cluster that have the same nucleotide
at every polymorphic site. For haplotype reconstruction, the similarity between
a candidate sequence and a haplotype group at each single SNP is calculated and
compared with a threshold to determine whether the candidate sequence matches
the haplotype group at that SNP; then the similarity over all SNPs is compared
with a second threshold to determine whether the candidate sequence can be
reliably assigned to the haplotype group. By using the similarity per
polymorphic site as well as the similarity over all polymorphic sites,
haplotypes can even be reconstructed reliably with sequences with sequencing
errors.
Thresholds for similarity per polymorphic site and
similarity over all polymorphic sites can be set according to the (suspected)
similarity between a gene and its paralogs, and the similarity between its
alleles. Too low settings for these thresholds may result in several alleles or
even paralogous sequences being classified as a single haplotype, while too
high settings would result in the separation of allelic sequences into
different haplotypes. After a series of tests on the potato data set, the
threshold value of similarity per polymorphic site is 75% and that of
similarity over all polymorphic sites is 80% are optional.
f. Settings for low quality
region
One of the criteria for identifying reliable SNPs is
confidence score, which is calculated by sequence redundancy in high quality
and low quality region. The extent of the low-quality regions at the 5' and 3'
ends of the sequences can be specified. Based on examination of public EST
sequences, we found that the 5' low-quality region is around 30 nucleotides,
while the 3' low-quality region is about 20% of the sequence length; these
values are set as defaults in HaploSNPer. The weight value for low quality
region can be set based on the quality of nucleotides in this region. In our
study, we found nucleotides in low quality region only have 0.5 of the
reliability. It means in low quality region at least two sequences have the
same nucleotide at a certain loci, and then the nucleotide is reliable. 0.5 is
the default for the weight value of low quality region. The value can be set by
users, for instance: if users don't want to use low quality region of
sequences, just set the weight value to 0.
g. Settings for SNP detection
In our program, sequence redundancy is also used to
prevent sequencing errors. The default setting for the minimum cluster size is
4, and the minimum allele size is 2. The default setting for the minimum value
of confidence score is 2. The confidence score is higher; the reliability of
the SNP on sequence redundancy is higher.
Based on the high quality (HQ) and low quality (LQ)
region, the confidence score of each allele is calculated according to the
score rules as defined in the following; and then the SNP confidence score is the
smaller one of each allele confidence score. The score rules for confidence
scores of each allele are as follows: 5 if the allele occurs in more than one
HQ; 4 if in one HQ and at least two LQ; 3 if in more than 3 LQ; 2 if in one HQ
and one LQ, or in 3 LQ; 1 if in 2 LQ, otherwise 0. In our study, we assigned a
high confidence score (HCS) to SNPs with a confidence score of at least 2. This
threshold can be adjusted by users according to their specific requirements.

h. Settings
for output
The output of HaploSNPer can be displayed in two ways:
the user can choose to view the cluster with the seed sequence, or all related
clusters.
An email address is required if users want the output returned to a
email address instead of waiting for a web page. As well, users can run a job
directly, and then it goes into a interactive process.
When the total number of homologous sequences (sequences users submitted and
sequences obtained from the selected database) is more than the threshold (100
sequences for human and mouse,and 200 for other
species), a email is required to send results back since it may take long time
to process. When the number of sequences is not more than the threshold,
HaploSNPer will show the results in a new webpage to the user directly.
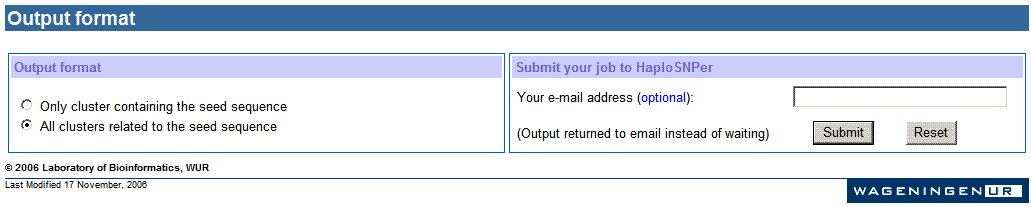
Note:
All these parameters can be set according to user's
requirements. Stringent settings will increase the number of reliable
haplotypes, and probably some false positive SNPs will be included. Loose
settings will decrease the number of reliable SNPs and some positive SNPs will
be missed.
Suggestion: Use the default settings to run
HaploSNPer, and then users adjust these parameters based on the results and
their experience.
3) The process of the program
After clicking the "submit" button, the
program will process the input sequences based on these settings.
In order to illustrate the performance of HaploSNPer,
sequences from the Mal d 1 genes of apple (Gao et al., 2005; TAG) were
used as the input. In this case the input consisted of a seed sequence and four
homologous sequences; and all parameters were kept as default. After submitting
the job, the processing page was showed as following:
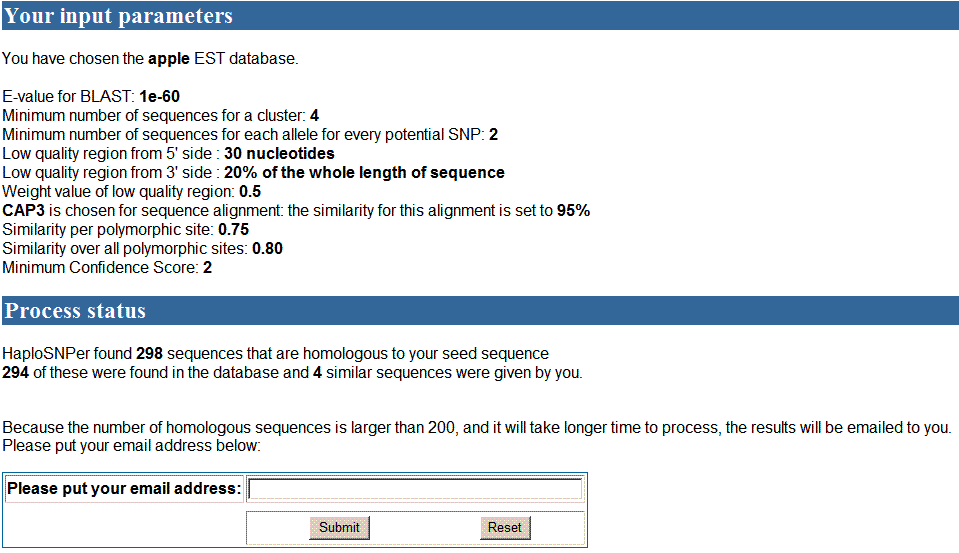
In the processing page, the database and parameters set
by user were showed. 294 homologous sequences obtained from apple EST database
and 4 similar sequences from the input that were more than 200 sequences in
total. An email address is required to send the results back when SNP detection
is finished since it may take long time to process them. Users put their email
address there and then wait for email. In the email, the link of results will
be contained. Users can read results by clicking those links.
If users want to get results directly, user can try to
lower E-value threshold. For example, E-value was set to e-180, suppose it
would get fewer sequences than 200, and other parameters still kept in default.
The processing page is as following:

193 sequences were obtained from database under the
threshold of E-value e-180. These sequences were merged with sequences (4 other
similar sequences and 1 seed sequence ) that the user
submitted were used to detect SNPs. There were two buttons showed in the page,
one button for showing results and the other for downloading results.
Note:
The results would be kept on our server in two days.
3. How to use the
results
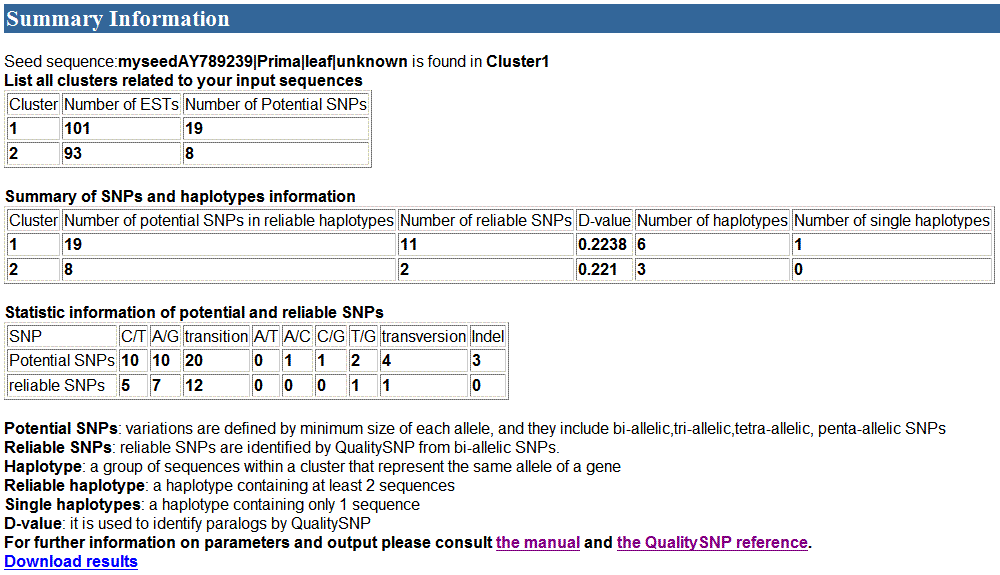
The results include three parts: the first part shows
parameters set for this process; the second shows the summary of haplotypes,
SNPs and D-value of all related clusters; the third part contains the haplotype
and SNPs information of each cluster.
1) The first part of the
results is as following:

2) The second part of the
results
3) The third part of the
results
If the output option "only the cluster with seed
sequence" is selected, only the cluster with the seed sequence will be
showed; If "All clusters related to the seed" is chosen, all clusters
with reliable SNPs will be showed one by one. The information for each cluster
insists of three parts (A, B and C). The A part is all information about the
potential SNPs and reliability of SNPs in the cluster. B part shows the
haplotypes reconstruction based on potential SNPs and sequence alignment
information by clicking the link "show sequence alignment
information". C part shows the reliable haplotypes and reliable SNPs. In
this case, two clusters were produced. In here the cluster with the seed was
used as an example. A part of the cluster follows:
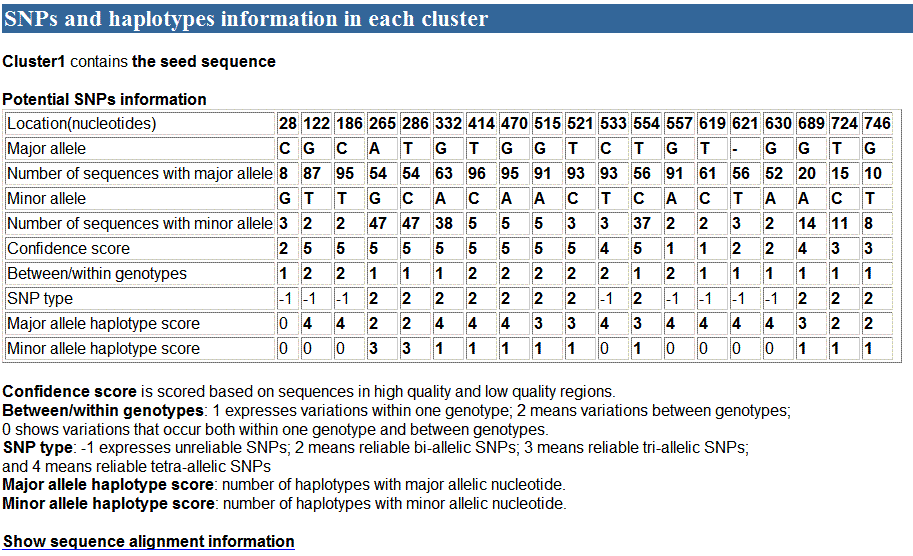
When click the link "Show sequence alignment
information", the sequence alignment and haplotype reconstruction (B part)
will be showed in a new window as following: 
In this window, the mouse is over the link of a
sequence accession, the annotation information (cultivar, tissue and unigene
ID) of the sequence will be showed, such as EB177853.
C part of the result focuses on showing reliable SNPs
and reliable Haplotypes, see the below.
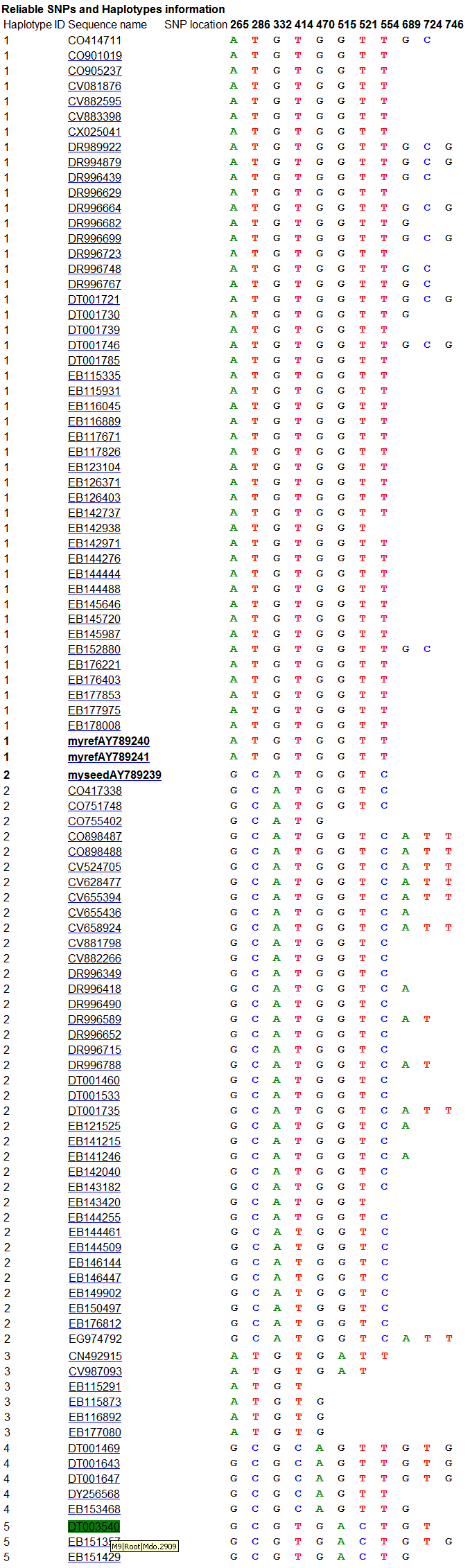
In A part, there were 19 potential SNPs defined in 5
reliable haplotypes, of which 11 were identified as reliable SNPs. In B part,
the haplotypes from 1 to 5 consist of at least two sequences, and were considered
as reliable haplotypes. In contrast, the last haplotype which consisted of one
sequence only, are considered as unreliable due to poor reliability of sequence
quality. These haplotypes would not be used to detect reliable SNPs. C part
shows relationship between reliable haplotypes and reliable SNPs.