|
In
the Simple Cloning Lab, PCR is used for 2 reasons:
-
To generate a high amount of DNA containing a particular region of
the Lambda DNA. This is done by PCR – amplifying the Lambda insert
of the pGTλ clone harbouring a recombinant plasmid.
After labelling,
the PCR products can be used as probes for Southern hybridisation.
-
To find out the size of the Lambda insert in pGTλ clones: the size of
the PCR product and the insert size are virtually the same
example: to discriminate colonies harbouring the recombinant plasmid
pGTλ3758 from those harbouring
pGTλ3758ΔH, PCR
with standard Gene Tech primers (see SCLResources>Sequences) can be used.
In
both cases primers specific for the pGT4 vector sequence are used. These
“standard Gene Tech PCR primers” anneal very close to the BamHI and
EcoRI insert sites.
The polymerase chain reaction (PCR) is an in vitro method for the
enzymatic synthesis of specific pieces of (target) DNA. It is a rapid
and simple means of producing (up to)
mg
amounts of DNA from minute quantities of target (“DNA amplification by
PCR”).
The
reaction requires:
-
two oligonucleotide primers that hybridize to opposite strands of
the target DNA and flank the region to be amplified
-
a
suitable DNA polymerase
-
the four deoxyribonucleoside triphosphates (dNTPs)
-
Mg2+
ions
To
perform a PCR amplification, a mixture containing the target DNA,
primers, dNTPs, and a heat-stable DNA polymerase is heated to 90-95°C
to denature the strands of the target DNA. The solution is cooled to a
temperature that allows the primers (single-stranded DNA molecules of
about 18 to 30 nucleotides long) to anneal to their complementary
sequence on the target DNA and provide the 3'-OH required for DNA
synthesis. Subsequently, the DNA polymerase synthesizes a new DNA strand
complementary to the target by extending the primer.
The polymerase used should be heat stable to tolerate the
high temperature denaturation steps of all reaction cycles. Such an
enzyme can be isolated from thermophilic bacteria like Thermus
aquaticus. The enzyme from this bacterium is most frequently
used in PCR. It is called Taq polymerase and has an optimal
activity at 72°C.
This thermal cycling scheme of -denaturing/primer annealing/ primer
extension- is repeated numerous times with the DNA synthesized during
the previous cycles serving as a template for each subsequent cycle.
The result is a doubling of the target DNA present with each cycle.
This exponential accumulation of DNA sequences can theoretically produce
over a millionfold amplification of the target in 20 cycles. In
practice, the amplification efficiency is less than 100%, and 20 to 40
cycles are commonly done.
The PCR is performed in a small volume
(20-100μl),
in small 200μl
tubes, or in multiwell plates. A heating block with an automatic thermal
cycler is used for precise temperature control.
As
little as one molecule of starting template can be enough for
amplification by PCR, because of the extreme amplification achievable.
Therefore, any source of DNA that provides one or more target molecules
can in principle be used as a template for PCR. This includes DNA
prepared from blood, sperm or any other tissue, from older forensic
specimens, from ancient biological samples or in the laboratory from
bacterial colonies or phage plaques as well as purified DNA.
New applications of the PCR are being developed constantly.
Whatever the source of template DNA, PCR can only be applied if some
sequence information is known so that primers can be designed.
Protocols for PCR vary considerably for particular applications.
In
the laboratory, “colony PCR” is often done: the reaction mixture is
set-up using intact bacteria picked from a colony on an agar plate,
rather than purified template DNA. The first denaturing step results in
release of the DNA from the (lysed) bacteria. Then the DNA is available
for annealing of the primers.
Primer length and sequence are of critical importance in designing the
parameters of a successful amplification: the melting temperature Tm
of a DNA duplex increases both with its length, and with increasing
(G+C) content:
a
simple formula for calculation of the Tm is
Tm
= 4(G + C) + 2(A + T)oC.
This formula could be used for primers smaller than 18 nucleotides
((G + C) means the number of C's plus the numbers of G's in the primer
sequence).
A
more accurate calculation of the Tm is according to the so-called
Nearest Neighbour methode. you could use
the
Oligonucleotide Properties Calculator
at
http://www.basic.northwestern.edu/biotools/oligocalc.html.
This type of calculation must be used for primers of 18 nucleotides and
longer.
Thus, the annealing temperature chosen for a PCR depends directly on
length and composition of the primer(s). One should aim at using an
annealing temperature Ta not more than 5oC below the
lowest Tm of the pair of primers to be used. Often, primers are
designed to have a Tm of about 60 oC
One consequence of having too low a Ta is that one or both primers will
anneal to sequences other than the true target, as internal single-base
mismatches or partial annealing may be tolerated: this can lead to
"non-specific" amplification and consequent reduction in yield of the
desired product, if the 3'-most base is paired with a target.
Read also about Hot Start PCR to avoid this.
A simple
set
of rules for primer sequence design
is as follows
-
primers should be 17-30 bases in length;
-
base composition should be 50-60% (G+C);
-
primers should end (3') in a G or C, or CG or GC: this prevents
"breathing" of ends and increases efficiency of priming;
-
Tms between 55-80oC are preferred;
-
runs of three or more Cs or Gs at the 3'-ends of primers may promote
mispriming at G or C-rich sequences (because of stability of
annealing), and should be avoided;
- 3'-ends of
primers should not be complementary (ie. base pair), as otherwise
primer-dimers can be synthesised preferentially to any other
product;
In the picture below the 3' end of
primer A is complementary to the 3'
end of primer B.
-
primer self-complementarity (ability to form secondary structures
such as hairpins) should be avoided. See
primer C in the picture below.
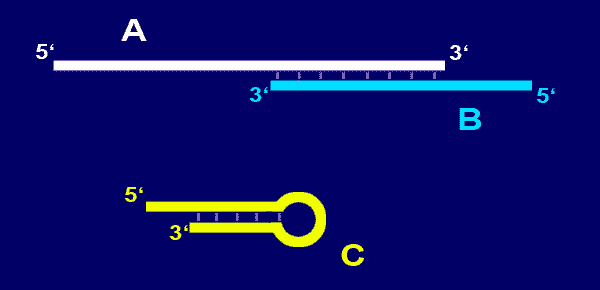
Primer-dimers are caused when two primers with the same or different
sequences hybridise. Usually this is found in PCR reactions where the
forward and reverse PCR primers hybridise. This appears as a 25-60 base
pair long product. In the picture below, primer-dimers formed by primers
A
and
B
are shown; the newly synthesized DNA in red.

Hot Start PCR
Hot Start PCR allows the inhibition of polymerase activity during PCR
reaction preparation. By limiting polymerase activity prior to PCR
cycling, Hot Start PCR reduces non-specific amplification and increases
PCR product target yield.
PCR
or polymerase chain reaction uses DNA polymerases which were isolated
from thermostable organisms. Unfortunately, these thermophilic DNA
polymerases show a very small but measurable polymerase activity at room
temperature during assembly of the experiment.
This in-efficient DNA polymerase activity will catalyze the extension of
any annealed 3' ends (so not only of 3’ ends of primers annealed at
their specific fully complementary target site..). After PCR
amplification, the resulting product contains a mixture of specific and
non-specific bands.
This
problem can be largely avoided by a very simple way: set up the PCR
experiment on ice, and wait untill the heating block is over 90°C,
before quickly transferring the PCR reaction tubes directly from ice
into the block.
This is the simplest way of performing a HotStart.
Better protocols for Hot Start PCR (i.e. absolutely no DNA polymerase activity during set up of the
PCR reaction) include chemical
modifications of the polymerase, wax-barrier methods, and inhibition by
an antibody against the heat-stable DNA polymerase.
PCR can amplify very small
amounts (e.g. a single copy) of template DNA.
So, very small amounts of contaminating DNA can give unexpected PCR
products.
Often, the setting-up of a PCR includes the preparation of a so-called
MasterMix, which is a large volume of a mixture containing all or
most of the PCR reaction components (buffer, dNTP's, primers,
polymerase), but without the DNA template.
Addition of the same mastermix to a series of different
template-containing samples makes it possible to accurately compare the
results of the samples.
One of the PCR reactions should be done without any template.
Instead of a template-containing sample, just water is used. This
control PCR is often called the water control.
It is very important to exclude in this way the presence of any template
DNA in (one of the components of) the mastermix.
When the mastermix is added to template-containing
samples which are already in the PCR tubes, it is extremely important to
use a fresh pipet tip for each addition!!
About the PCR machine
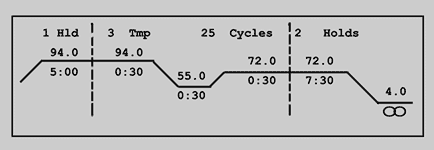
The picture above is typically shown in
the display of a PCR machine, during (setting-up) a PCR run. You
should read it like this:
There is a first denaturation step of 5 minutes at 94°C, followed by 25
identical cycles, with in each cycle a denaturation step of 30 seconds
at 94°C, a primer annealing step of 30 seconds at 55°C and a primer
elongation (by the polymerase) step of 30 seconds at 72°C. Finally a 7.5
minutes elongation step at 72°C has been programmed, followed by cooling
to 4°C until the user stops the run manually.
|